So, this is my first ever blog post (both work and personal) and I wanted to use this to talk about differing dose escalation (DE) practices, especially between first in human (FIH) healthy volunteer trials and those in patients (FIP). In reality, if you are complying with Good Clinical Practice (GCP) they should be the same.
More frequently, with the advancement of medicine, FIH trials are being conducted in patients. Biologicals, cell therapy or oncology medicines may be too toxic or not suitable to give to healthy volunteers, so patients become the first humans to receive these medicines.
Over the years FIH trials have had an excellent safety record, with two recent well-publicised exceptions: the dosing of TGN1412 in London in March 2006 (the day I started at the agency as a GCP Inspector) where 6 volunteers became extremely ill, and more recently, in January 2016, the dosing of BIA10-2474 in France, where one volunteer died and four others became seriously ill.
The TGN1412 event resulted in FIH healthy volunteer trials and their conduct being scrutinised, and various early phase guidance issued or updated1. In the UK it also led to the implementation of the voluntary phase I accreditation scheme in 2007, with the first unit accredited in April 2008. (The scheme currently has 14 accredited units - 10 commercial and 4 non-commercial. Information on the scheme and the accredited units can be found on the MHRA website). However, even with this guidance a second event occurred in France, resulting in further scrutiny and updating of the EMA guidance, which was adopted in July 2017.
Despite these events, early phase and FIH trials remain remarkably safe due to the scrutiny of well-developed protocols and supporting regulatory documents for the medicines tested. But sponsors, clinical research organisations (CROs) and regulators still have a continuing duty to ensure that risks are minimised for all types of FIH trials, including patient trials, and that GCP procedures to minimise and mitigate those risks are followed. European (EU) Directives 2001/20/EC (the Clinical Trials Directive) and 2005/28/EC (the GCP Directive), which were implemented into national law in all EU member states, enshrine the GCP principles into law. (In the UK these are the Statutory Instruments SI2004/1031 and 2006/1928).
Having inspected many phase I and FIH trials over the years as part of general GCP inspections, as well as the phase I accreditation scheme assessments, one area where I and other MHRA colleagues see differences in is DE. I want to talk about these differences and why they occur - but also why they should not be occurring.

While we may not have had any disasters that we know of in the EU in patient FIH trials, we need to apply the same level of risk mitigation to this population as we do to healthy volunteers. The reason I say ‘know of’ is that patient trials usually involve seriously ill patients with added complexities. Therefore, any toxicity resulting from the new medicines may be masked for several reasons (such as severity of the illness, drug interaction or combinations). However, this is not a reason to reduce the risk mitigation or compliance with GCP as there are two US examples of phase I gene therapy trials that have resulted in patient deaths.
In 1999 a US patient recruited to a phase I gene therapy trial was injected with an adenoviral vector carrying a corrected gene to test the safety of the procedure. He died four days later, apparently having suffered a massive immune response leading to multiple organ failure and brain death. An investigation by the Food and Drug Administration (FDA) identified a failure of the sponsor to report previous patients who had experienced serious side effects from the gene therapy.
Also, in 2017 two US phase I gene therapy trials were halted following the death in one trial of a patient from severe cytokine release syndrome and the serious illness of a second patient with similar side effects.
It has long been established in FIH healthy volunteer trials that dose escalation is a thorough, detailed and well documented process. It involves the sponsor and the principal investigator(s) (PI) making decisions based on sound data. However, this practice does not seem to transfer to the multi-centre patient trials, even for the same sponsor, whether they are commercial or academic/non-commercial.
Why?
![]()
And why is it so hard to get sponsors to understand that these differences are not appropriate - or for them to take advice from accredited units who have both a wealth of experience in this area and effective procedures in place to manage the process?
The history of these differences may lie partly in the fact that traditionally healthy volunteer trials were conducted in a single specialised unit with a PI experienced in pharmacological development. Patient trials, on the other hand, would be conducted as multi-centred trials by a number of therapeutic specialist PIs relying on the sponsor for the pharmacological experience. Changes in technology and data collection also play a role.
So, what are the GCP expectations for any dose escalation trial?

Let’s start by referring to some crucial GCP requirements specified in the EU directives and how I interpret these.
| EU Directive | Article | Statement | Interpretation |
| 2001/20/EC | 1(2) | Good clinical practice (GCP) is a set of internationally recognised ethical and scientific quality requirements which must be observed for designing, conducting, recording and reporting clinical trials that involve the participation of human subjects. Compliance with this good practice provides assurance that the rights, safety and well-being of trial subjects are protected, and that the results of the clinical trials are credible. | The sponsor, Data and Safety Monitoring Board (DSMB) (or any other committee) and the investigator must follow the GCP principles as set out in the directives and ICH GCP E6. |
| 2001/20/EC | 1(4) | All clinical trials, including bioavailability and bioequivalence studies, shall be designed, conducted and reported in accordance with the principles of GCP. | |
| 2001/20/EC | 3(2a) | Before the trial is initiated, foreseeable risks and inconveniences have been weighed against the anticipated benefit for the individual trial subject and other present and future patients. A trial should be initiated and continued only if the anticipated benefits justify the risks. | A thorough and documented risk assessment and mitigation plan, especially covering items not detailed in the protocol (such as logistical aspect of DE). |
| 2001/20/EC | 3(3) | The medical care given to, and medical decisions made on behalf of, subjects shall always be the responsibility of an appropriately qualified doctor or, when appropriate, of a qualified dentist. | The investigator has the medical responsibility for the trial subjects under their care. Ensuring that they have the information about the data used (for example, for starting dose and for the DE decision) so they can make the decision for the subjects under their care. |
| 2005/28/EC | 2(1) | The rights, safety and well-being of the trial subjects shall prevail over the interests of science and society. | Safety of trial subjects is protected by ensuring that data used is accurate (such as data for DE) and that the PI is satisfied with the DE decision. Verifying the accuracy of the data (QC) can confirm this. |
| 2005/28/EC | 2(4) | The necessary procedures to secure the quality of every aspect of the trial shall be complied with. | Procedures (for example, QC) ensuring the quality of data used for DE decisions. |
| 2005/28/EC | 3 | The available non-clinical and clinical information on an investigational medicinal product shall be adequate to support the proposed clinical trial. | All data used for the design of the trial and any decisions made during the trial (such as DE data) shall be of a suitable quality for strong decision making (such as QC for accuracy and completeness). |
| 2005/28/EC | 4 | The investigator and sponsor shall consider all relevant guidance with respect to commencing and conducting a clinical trial. | In the UK, sponsors/PIs should also take into consideration the MHRA phase 1 accreditation scheme and guidance (version 3) plus The Association of the British Pharmaceutical Industry (ABPI) guidelines for Phase I trials (edition 2018) in addition to EMEA/CHMP/SWP/28367/07 (EMA guidance on identifying and mitigating risk in FIH trials). |
| 2005/28/EC | 5 | All clinical information shall be recorded, handled and stored in such a way that it can be accurately reported, interpreted and verified, while the confidentiality of records of the trial subjects remains protected. | Any data including DE data should be accurately reported and verified – therefore QC is required. |
| 2005/28/EC | 16 | The documentation referred to Article 15(5) of Directive 2001/20/EC as the trial master file shall consist of essential documents, which enable both the conduct of a clinical trial and the quality of the data produced to be evaluated. Those documents shall show whether the investigator and the sponsor have complied with the principles and guidelines of GCP and … | The trial master file (TMF) (which comprises of the sponsor and investigator files) shall retain evidence to be able to reconstruct the DE process, including the collection, QC, and circulation of the data and the decisions. |
| UK Specific regulation | |||
| SI2004/1031 as amended |
51 (1) | Defence of due diligence:A person does not commit an offence under these Regulations if he took all reasonable precautions and exercised all due diligence to avoid the commission of that offence. | A subject experiences harm from participation in a trial due to decisions made on inaccurate data. If the PI had not exercised any due diligence to satisfy themselves that all the data had been QC’d and accurate and not seen all the data used for the decision, then this would prohibit the investigator from using this defence and they could be prosecuted. |
Phew, now we’ve got all that regulatory stuff (which I know some of you might find boring) out of the way, why am I bringing this to your attention? Well….
Although there should be no need to spell this out, when clinical trials are authorised by the regulator and approved by the ethics committee, they are expected to comply with the regulations, the protocol, GCP and any associated early phase guidance1. Deciding to place patients or healthy volunteers on a higher dose is a safety decision and therefore should be based on accurate information.
![]()
The MHRA phase I accreditation scheme has ingrained into accredited units the importance of defining the DE procedure transparently in the protocol but this does not seem to have been adequately considered by the sponsor (both commercial and academic/non-commercial) especially for multi-centre trials. So, the expectation is that unless the protocol defines otherwise (for example, it specifies a percentage of the subjects, a specific set of data or a maximum timepoint) then all the data from all the subjects in the cohort will be used for the dose escalation meeting and decision.
An MHRA CTU medic confirmed “We would always expect that the protocol defines an absolute minimum for review in terms of numbers of subjects and data to be reviewed”.
Also, it is expected that accuracy of data will be ensured via quality control (QC) processes (either by the unit or by sponsor monitoring) especially for data linked to major safety and stopping criteria including pharmacokinetic (PK) or pharmacodynamic (PD) data (for example, a Cmax or AUC).
There should be a DE process where the PI(s) and the sponsor review all the data and make a documented decision. This meeting, together with the data used (including evidence of QC) and the decision, should be clearly documented and the decision circulated to all relevant parties (such as clinical and pharmacy teams) in advance of the next dosing occasion. An MHRA CTU medic stated, “We also expect that a summary of the communication plan is in the protocol”.
So, for DE to be GCP-compliant, there should be a formalised procedure detailing how the dose escalation process will be undertaken (in the form of SOPs and transparency in the protocol).
Why is a dose escalation process important?

Well, my colleagues and I have identified DE issues. Here are some examples:
- no procedures for how DE will be conducted in the protocol or separate document (such as standard operating procedure (SOP) charter/study specific plan)
- a lack of documented evidence of quality control (QC) of the safety data listings provided to the DE committee resulting in poor quality data used for the DE decision:
- serious adverse events (SAEs)/adverse events (AEs) not transcribed/ missed that were contained in the source data
- SAEs/AEs had no causality/severity assigned
- safety data missing from the data listings (such as important safety laboratory results and electrocardiogram (ECG) data)
- PK data and other data outlined in the protocol for review was not present in the data provided to the DE committee
- the SOP for collation of interim safety data for data monitoring committees, which included DE, clearly stated the use of ‘dirty’ or ‘unclean’ data but these documents were not seen by the regulator or ethics committee approving the trial
These issues have either resulted in the approved protocol stopping criteria being missed - with DE proceeding when it should have stopped - or had the significant potential of being missed. A decision to continue to dose escalate increases risks to the safety of the next cohort of subjects. A repeat or smaller dose increase decision may have been taken if the missing/more accurate data had been present.
Have dose escalation issues always happened or is this a new phenomenon?
I think it could be a combination of both. ‘Why?’ you ask. Well here are some potential reasons:
- DE for healthy volunteers routinely happened at a single site in the past, so the PI knew all the subjects recruited and had control of all their data. Now, we’re seeing more multi-centre trials (for healthy volunteers and patients). This means the DE data is outside the control of the PI and at the sponsor/CRO level so the PI cannot verify the quality of all the data collected, only the sponsor/CRO can do this. As a result, accredited units are not able to easily comply with the accreditation scheme requirements, which require GCP compliance for DE
- The use of electronic case report forms (eCRFs) means the sponsor has the data sooner, but it has not necessarily been monitored/source data verified (SDV’d) to confirm accuracy. Previously, with paper CRFs, they would have been SDV’d by the monitor before being collected for data entry, so a QC had been performed. Whilst technology has allowed the data to be accessed earlier, sponsor/CRO procedures identifying the use of ‘dirty’ or ‘unclean’ data, have not been revisited - these previously described data entered into the database from SDV’d paper CRFs, not unmonitored eCRF data. So, you might say ‘dirty’ data has just got a whole lot dirtier!
- Investigator sites are busier and time constraints mean eCRFs may not be updated in time for DE meetings
- With the increase in multiple vendors collecting various data used for DE decisions (for example, cardiology/ECGs, different laboratories for safety, PK and PD) all feeding into the DE data listings, vendors are not always aware of the use of their data and the importance of ensuring an adequate level of QC prior to providing the data. Also, sponsors/CROs are not routinely ensuring that all parties are providing complete and accurate data.
Where does that leave us then?
We’re all in agreement that in today's clinical environment we should be thinking about quality by design.
We also need to be looking in a pragmatic and risk-proportionate way to the trial objectives, the population being recruited, the various sources of data and how this is being collected and used. So, to comply with GCP in DE, the starting point is with a risk assessment and mitigation strategy:
- Are all the subjects in the cohort needed to make a dose escalation decision? Yes or no?
If no, is this described with a clear justification in the protocol? Because it should be, otherwise the regulator will expect all subjects to be included. Also, think about what the protocol says about replacing withdrawn subjects, as this could inadvertently reduce the size of your cohort. - Is all the data from all the timepoints in the cohort needed to make a dose escalation decision? Yes or no?
If no, are the key assessments and the minimum timepoints needed described with a clear justification in the protocol? Because it should be, otherwise the regulator will expect all the data from all the timepoints. - What are the stopping criteria and what is being stopped? An individual? A cohort? The whole trial?
This should be clearly defined in the protocol. - Does the minimum data and timepoints required ensure that the data used covers all the stopping criteria defined in the protocol?
- What is the quality of the data used to make a dose escalation decision? Will the data be verified for accuracy in some way? Yes or no?
If no, can you provide assurance that the data is reliable and accurate enough to be used to make a crucial safety decision in the trial? Consider what data is really needed (for example, that associated with stopping criteria) and consider if there are justifications for reducing the GCP expectations for reliable data. There may be some level of QC or SDV that can be performed on important data. Or, in some cases, there may be a justifiable rationale for reduced QC for the DE data (such as patients where all lines of conventional treatment are exhausted, and this is their last hope, so dosing time is of essence). If this is the case, this should be clearly described in the protocol so the regulators can see your rationale and be aware of any reduced QC in advance. - Have you considered where all the data is coming from? If the data does need to be verified for accuracy, do study-specific plans and any vendor-applicable contracts include the requirement to QC that data?
So, what’s my take home message?

In summary:
- The MHRA Clinical Trials Unit (CTU) approving the trial protocol will assume a GCP-compliant DE plan, meaning that all subjects in the cohort and all their data at all the timepoints will be reviewed in the DE decision meeting unless the protocol states otherwise
- The MHRA GCP inspectors inspect against the legislation and the relevant early phase guidance1 so expect to see a formalised procedure for DE and for a specific clinical trial to be able to reconstruct the dose escalation as described in the formalised procedure and approved protocol. This will verify that the trial was conducted in a GCP-compliant manner. In summary, we will expect to see:
- evidence that the relevant data has been collated, verified for accuracy and provided to the DE committee
- the DE meeting minutes
- the DE decision agreed by the DE committee (including the PIs)
- circulation of the DE decision to all relevant team members
It’s important to remember that the central focus for phase 1 trials is safety, so this post does not just apply to the phase I accreditation scheme but to anyone conducting these types of trials.
Also, in today’s complex trial design and expansion into the patient first in human trials, have you really considered if your current procedures are in line with new technology for collecting and using accurate and reliable data for safety decisions?
Have you built quality by design into the protocol, risk assessed the trial design and ensured your decisions are transparent to the regulators and ethics committees reviewing the protocol?
Finally, does the trial master file enable you to reconstruct the dose escalation process easily? (See below)
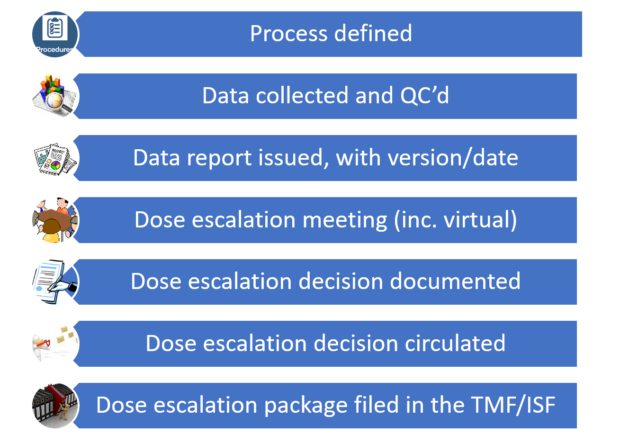
Remember, the Agency’s primary objective is to protect public health and to ensure the quality of data. Dose escalation is a major safety decision in a trial so good quality data is vital to ensure the safety of the subjects.
We’re Here to Help
Dose escalation queries for both MHRA CTU assessors and GCP inspectors can be directed to ctdhelpline@mhra.gsi.gov.uk
1Early Phase Guidance:
- EMA guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products
- ABPI Guidelines for Phase I clinical trials
- UK MHRA Phase I Accreditation Scheme & Guidance
- MHRA GCP Guide
Don’t miss the next post, sign up to be notified by email when a new post comes out


3 comments
Comment by Thuy Larsen posted on
Great and informative article!!! Thank you for sharing your opinion on this important topic. Highly appreciated ;=)
Comment by C. Thorpe posted on
Yes, this is very important information to help standardize studies. Thank you!
Comment by Siobhan McGrath posted on
Jennifer
Excellent article with clear and concrete advice. I have drawn the attention of this article to the Chairing teams of the HSC Research Ethics Committees in Northern Ireland
Dr Siobhan McGrath (Head of the Office for Research Ethics Committees Northern Ireland, ORECNI at BSO)