
We all know it’s important to ensure we have accurate and consistent data across all our regulatory documents for clinical trials. We also want to ensure that high risk trials are appropriately risk-assessed by the sponsor and therefore authorised and approved under the right categories by the regulator and ethics committee.
So, before you complete those all-important Clinical Trial Application (CTA) and Integrated Research Application System (IRAS) forms, are you ensuring you have appropriately identified/categorised the type of trial and therefore correctly identified and mitigated all the risks associated with that type of trial? That way you will ensure the forms have the correct information.
Let’s take the type and phase of a trial:
- CTA section E7 covers ‘TRIAL TYPE AND PHASE’: E7.1 asks ‘Human pharmacology (Phase 1)?’ Yes/No. If yes, E7.1.1 asks, ‘Is it First administration to humans?’ Yes/No
- IRAS question 2(a) asks, ‘Is this a commercially sponsored Phase 1 or Phase 1/2a trial involving healthy volunteers?’ Yes/No. It then asks in question A9-1 ‘Is it a Human pharmacology (Phase 1)?’ Yes/No. Finally, it asks in 15-3 ‘Is it an IMP to be used in a first-in-human clinical trial?’ Yes/No/not answered
While in the long run it may not impact on the authorisation decision by the MHRA Clinical Trials Unit (CTU) or approval by the ethics committee, it can impact on:
- how you identify and mitigate the risks associated with the clinical trial
- how your clinical trial will be inspected by the MHRA GCP inspectors in relation to GCP and early phase guidance (for example, EMA, ABPI and MHRA voluntary phase 1 accreditation guidance1)
- meeting GCP requirements for trial activities, such as dose escalation
- expectations for accredited phase 1 units maintaining their accreditation (for example in meeting requirements for dose escalation and resourcing for medical emergencies)
So, thinking about how trials might be inspected and following on from that, you can see that when risk-assessing early phase clinical trials, it’s important to be able to identify the true first in human (FIH) trials from other early phase trials.
So, here are a couple of flowcharts with some questions you might want to ask yourself when you are doing your risk assessment and completing your IRAS and CTA forms:
Flow chart 1: Is it a ‘true’ FIH trial?
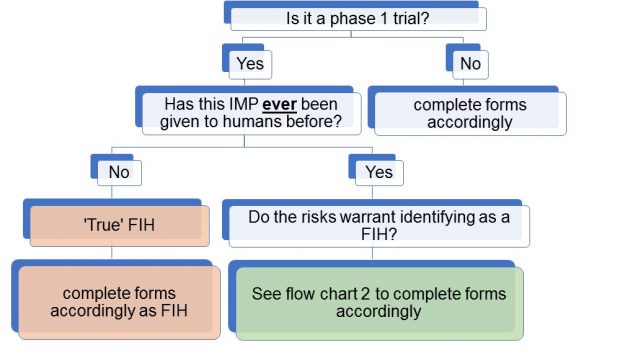
So, if it’s not a ‘true’ FIH trial, as the IMP has been given to humans before, then other factors may come into play which might influence the decision as to whether it should be considered a FIH trial.
The MHRA CTU considers that two licensed drugs used in novel combination or in a new therapeutic area are not first in human nor necessarily a phase 1 trial. Phase 1 guidance1 - including the MHRA voluntary phase 1 accreditation scheme - concentrates on pharmacology and toxicology. If a trial is classified as phase 1 and specifically as a FIH trial, the MHRA GCP inspectorate will inspect it as such.
The MHRA CTU does not take a view on how a sponsor classifies a study, so will not raise this as a comment or grounds for non-acceptance (GNA). We also acknowledge there is no standard definition beyond that of guidelines which state a phase 1 trial is not expected to have therapeutic benefit to the participants. An early phase trial does not have to be categorised as phase 1 if it is perceived to have benefits, although potential benefits may not be predictable if given to a new population or in a different combination.
Flow chart 2: For IMP that has been given to humans before, do the risks identified warrant categorising it as a FIH? (some examples)

So once, you’ve decided what type and phase of trial it is, you can confidently risk-assess your clinical trials and complete your CTA and IRAS forms. This will then allow MHRA GCP inspectors to inspect the clinical trial to the applicable legislation and guidance. It will also help inspectors and MHRA accredited phase 1 units to identify those trials appropriately for assessment against the MHRA phase 1 accreditation scheme. Other early phase trials may be selected to help the GCP inspectors assess a unit’s procedures but will be reviewed in the context of the type and phase of trial it has been recorded as in the CTA and IRAS forms.
So, as a sponsor, if you make the right decision about the type and phase of trial, it will make life easier for you and for all parties involved in the clinical trial to comply with the various legislation and early phase guidance, including the MHRA voluntary phase 1 accreditation requirements.
Remember, all early phase clinical trials have risks associated with them that require mitigation, thus following not just the legislation but also all the associated guidance will help in maintaining public safety and the credibility of data.
If in doubt, please discuss this with either the MHRA CTU or an accredited phase 1 unit, who will be able to provide expert advice. You can also find useful information in the EMA guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products, adopted in July 2017.
1Early Phase Guidance:
- EMA guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products
- ABPI Guidelines for phase I clinical trials
UK MHRA Phase 1 Accreditation Scheme & Guidance
Don’t miss the next post, sign up to be notified by email when a new post comes out


2 comments
Comment by Sam Mowbray posted on
Certainly an interesting read. In terms of the microdosing studies mentioned, as the studies following on from microdosing studies are to be considered FIH, would this therefore mean that any initial microdosing study (including PET radiolabelling studies etc) of a new molecule would themselves fall outside the scope of being classed as FIH (assuming the requirements of ICH M3 for microdosing studies were met), or would the process as outlined in flow chart 1 still apply? This would be a useful point of clarification for anyone involved in the setup of such studies.
Comment by Janet Symes posted on
Dear Sam
A microdosing CTIMP would still also be considered a FIH trial and all FIH principles would still be expected to be applied, unless justified. The microdosing study would be the true FIH with the follow-on studies also being considered as FIH in principle as the microdoses cannot be expected to provide any meaningful safety data. Thus the follow-on studies should be considered FIH as there will be no or little supporting clinical data.
Kind regards
Kirsty