Earlier this year I represented the MHRA at the 10th Workshop on Recent Issues in Bioanalysis (WRIB).
This is a large international conference attended by approximately 700 people from pharmaceutical companies, biotechnology companies, contract research organisations and colleagues from other regulatory agencies from around the world. The week was made up of three full days of presentations and panel discussions bracketed by a variety of training courses.

WRIB is an important conference to attend for both regulators and industry alike. From my perspective as an inspector it provides me with an opportunity to hear about advances in technology, new approaches to bioanalysis, topics where industry is having problems and feedback from other regulators on areas of interest and concern. The information gained can then be shared with my colleagues and used to inform our inspection approaches going forward.
The main workshop spanned three days, running between 8am and 6.30pm each day with 51 presentations, three formal panel sessions and the infamous ‘black screen’ if presenters happened to run over their allotted time. In addition, each evening the various speakers from industry and regulatory authorities are requested to meet for a working dinner to discuss a wide variety of topics which ultimately lead to the publication of a white paper covering the conclusions and consensus points of the conference (previous white papers are listed here.
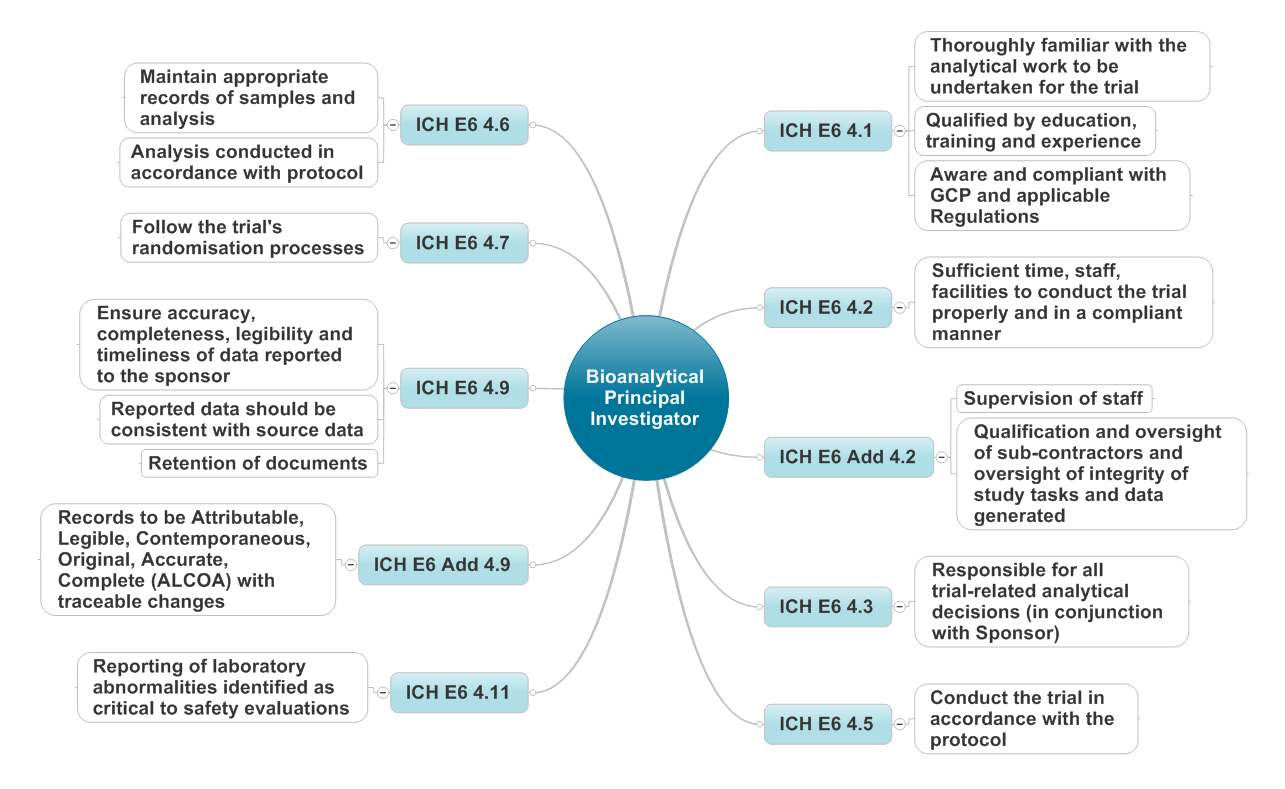
I gave a presentation on the role of the Bioanlytical Principal Investigator in relation to clinical trials (which I’ve also heard called Principal Scientist for Clinical Trials, Study Director and Laboratory Investigator to name a few). I suggested that the role of the Principal Investigator defined within ICH GCP can be almost directly mapped to the role of the Bioanlytical Principal Investigator “a person responsible for the conduct of the clinical trial at a trial site. If a trial is conducted by a team of individuals at a trial site, the investigator is the responsible leader of the team and may be called the principal investigator”. With a little tweaking of the wording of ICH GCP on the roles and responsibilities of a clinical investigator a good set of job descriptors emerges (see the diagram below) which apply to the bioanalytical principal investigator.
I also spoke about our expectations at the MHRA associated with unblinding of sample analysis results and suggested a number of points to consider before revealing the treatment allocation. There must be always be a valid reason to unblind the trial samples. The first question to ask is whether knowledge of the treatment allocation is likely to make a difference to the understanding of an analytical issue under consideration. If unblinding samples is the only way in which the information can be obtained then it is important to have a documented rationale clearly stating the decisions made and to have this approved by the trial sponsor. Once agreed, it then becomes a question of information provision and control. The bioanalytical principal investigator, in conjunction with the sponsor, needs to work out how to restrict the information to those staff who need it in order to progress the investigation of the analytical issue and how the unblinded information will be secured against unauthorised access. Unblinding outside of the strict end of trial processes always has the potential to become a data validity or integrity issue so care must be taken so that there are no ‘knock on’ effects on the rest of the trial.
I’d also like to mention some of the points raised during other presentations and during the Regulatory Agency panel session.
Stability Experiments
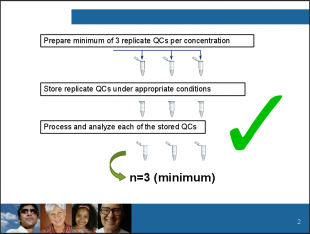
Dr Mark Bustard (Health Canada) spoke about a notice which had been issued by Health Canada in October 2015 (and an additional addendum in March 2016) associated with matrix-based stability experiments for bioavailability studies. Stability experiments are conducted to demonstrate that the analyte being measured is not adversely affected by the passage of time, the effects of temperature or any other factors which may impact on the quality of the analyte being measured. The EMA Guideline on Bioanalytical Method Validation describes the approach to be taken using QC samples which are analysed immediately after preparation and following exposure to the storage conditions being assessed.
A change in practice by industry had been identified which was deemed by Health Canada to be unacceptable. Mark described that historically summary results from multiple sample tubes (typically six) were reported but over time this practice has changed to one bulk sample being stressed and then aliquoted into six tubes for analysis. The results from this modified approach were then reported as having been from six tubes whereas this is actually a misrepresentation as the stability experiment was only conducted on one bulk sample tube. Mark then went on to explain that the longstanding expected approach is that a minimum of three replicate quality control sample tubes per concentration are to be prepared, that these samples are stored under appropriate conditions and then each of the stored tubes is processed, analysed and reported as n=3.
Analysis of Clinical Samples
The regulator panel was asked whether bioanalysis was to be conducted according to Good Laboratory Practice (GLP) or Good Clinical Practice (GCP). The expectations of the majority of global regulators are clear – analysis of clinical trial samples must be conducted according to GCP (although some regulators do specify GLP as a requirement). The position we follow in the UK is outlined in the UK GLP Regulations which clearly state that GLP is only applicable to pre-clinical activities. It is recognised that some aspects of GLP may be beneficial to the conduct and management of clinical sample analysis but, that by solely following GLP, issues that may impact on the safety and rights of subjects would not be addressed. Additional guidance for laboratories that perform the analysis of clinical trial samples can be found here.
Risk Adaptive Analysis
Finally there was discussion associated with risk adaptive approaches to bioanalysis in clinical trials. Concerns were shared by the regulators and audience alike about large amounts of blood being taken from subjects without a clear plan for the experiments and tests to be conducted as part of the trial, with a view to adapt the type of analysis as the trial progressed. The audience was reminded that the planned analysis strategies should be included within the protocol and that appropriate approval must be obtained from the relevant competent authority and ethics committee for the use of the samples. Additionally, informed consent must be obtained from the patient for the specific uses of their sample.
So it was a long and very busy week but a very worthwhile event to attend. I’d like to thank Dr Mark Bustard for permission to use his slide and also to the WRIB organisers for the invitation to attend and speak at the workshop.


Recent Comments