In this post you’ll find the first in a series of ‘frequently asked questions’ related to the updated EU GMP Annex 16 on QP Certification and Batch Release.
I have been a GMDP inspector since October 2014, and part of my role is to perform inspections of manufacturers, importers and batch certification sites.
I trust that you find the following Q&A’s helpful. If you have any further queries on this or other GMP subjects, please continue to send them to the GMP Inspectorate email address.
Sampling and testing on importation
-
How should the Pharmaceutical Quality System be used to support reliance on the vendor-supplied samples for importation purposes?
This could be similar to a typical vendor assurance programme for the qualification of a new vendor. For example, comparative analysis of samples taken in the third country and further samples taken after importation should be tested together, until a justification can be supported to rely on third country samples only. There should be assurances (through audit and technical agreement) that the manufacturing site takes the samples according to approved procedures, and that the samples are random and representative of the entire batch. For sterile products the sterility testing requirements would need to be carefully worked through to ensure the site of manufacture could react appropriately.
-
When does the comprehensive scientific study need to be in place?
The implementation date was 15th April 2016. For sites importing a large number of products (say 10 or more), it is recognised that the technical justification and comprehensive scientific study may not be in place for all products immediately. A plan to have all studies in place soon thereafter will be accepted. The site will need to demonstrate they have identified the high risk sites and have an action plan to ensure the studies are being worked on in an acceptable time frame and according to risk.
-
How and when should the comparative analysis of samples be performed?
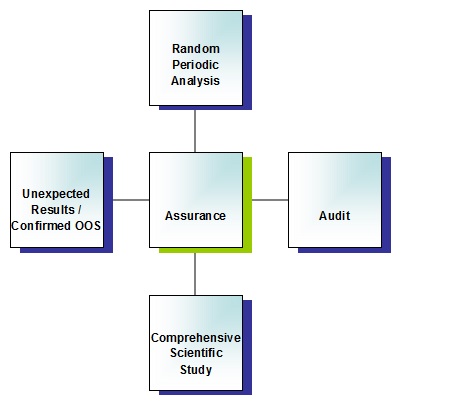
This refers to parallel QC testing of samples from the same batch; in other words, samples taken at the manufacturing site, and further samples taken at the site of importation. In both cases, the samples should be representative (i.e. taken from throughout the batch), and evidence of this demonstrated. Batches chosen for sampling in the EU for the periodic analysis should be random, and the third-country manufacturer should not know which batch is chosen prior to importation, sampling or testing.
Samples taken within UK WDA premises for the purpose of periodic analysis are to be whole packs only (as described by the marketing authorisation). No breakdown of a full pack is allowed. It is expected that the representative requirement for these samples will mean that multiple full packs are removed from the batch. Any such arrangements must be described in a technical agreement, the sampling should be included in the audit of the UK WDA holder by the manufacturer, and a copy of this audit made available.
The programme of analysis (i.e. the tests to be performed, and the frequency of the random periodic analysis) should be outputs from a risk assessment.
-
Can other member states WDA / authorised storage and distribution sites be used to store and sample the imported stock?
The UK decision to allow a WDA holder named on a UK MIA to store and periodically sample uncertified stock applies to UK sites only.
In other member states the local rules must apply.
-
Does the finished product sampling for EU testing at third country manufacturing sites need to be part of the scope in audit reports?
It is recognised that importers will still be reliant on legacy audit reports until the next scheduled audit. In such cases it is expected that a formal assessment should be performed, clearly stating whether the existing audit report covers sampling of the finished product for EU testing or not. Where the audit report does not cover this sampling, the importer should determine (and document in their formal assessment) whether there is any other supporting information. For example, is the sampling activity described in a technical agreement? Is the sampling procedure approved by the batch certification site?
Existing technical agreements should be reviewed to ensure that finished product sampling is included. Audit plans and related SOPs should be updated to include finished product sampling for EU testing at third country manufacturing sites. The assessment of audit frequencies should be assessed for continued suitability.
-
What is required by ‘a review of any unexpected result or confirmed out of specification result’?
It is assumed that a confirmed out-of-specification result will be obvious. An ‘unexpected result’ should be described in site procedures. Unexpected results from samples taken in the third country should be treated no differently to an unexpected result obtained by samples taken in the EU. Other unexpected results may include out-of-trend indicators, such as a shift in results or a change in profile.
The investigation should be thorough and complete before a conclusion is reached to establish which of the results is accurate. The review should include the wider compliance picture as a consequence of finding an OOS/OOT (for example, is this the only product manufactured by the site? what is the implication to other products/batches?).
The review should reach a conclusion on whether other batches are affected, whether market action is required, whether reliance of sampling in the third country can still be supported, and any implications for the third country manufacturing site (e.g. robustness of Pharmaceutical Quality System).
Where required, the notification to the supervisory authority is to be made by the MA holder or the site of batch certification, depending on the requirements of the technical agreement. The notification is to be timely and commensurate with the potential market action, as per EU GMP Chapter 8.
Integrity of imports
-
What is required for ‘Evidence should be available to ensure that the integrity and identity of the imported finished product batch has been established through documented verification’?
The evidence is to be made available to the QP at the site of batch certification.
The consignment should have remained secure, with no evidence of tampering during storage or transportation. There are a number of different ways of identifying if a shipment has remained integral. For example, the use of numbered or uniquely identified tags that are recorded both at the third country and by the receiving site, printed stretch seals/overwraps or printed closure systems, use of photographs, etc. The batch size, number of consignments/shipments and the shipment route should all be confirmed. Evidence of continued storage at the required temperature / moisture / light exposure levels should be documented.
The method of identification is to be decided by the importer/MA holder, based on the product requirements and the process constraints of the manufacturer. This can be visual inspection where no risks are envisaged. It may require a finished pack to be broken down into the components to confirm identity. In high risk scenarios it may require analysis.
Supply chain diagrams
-
What are the expectations for the documentation of supply chains?
The primary requirement is that the supply chain is clearly visible to the certifying QP. The entire supply chain of the active substance and medicinal product up to the stage of certification is to be documented. This is required for each product; though it could be grouped where appropriate, for example for different strengths of the same product.
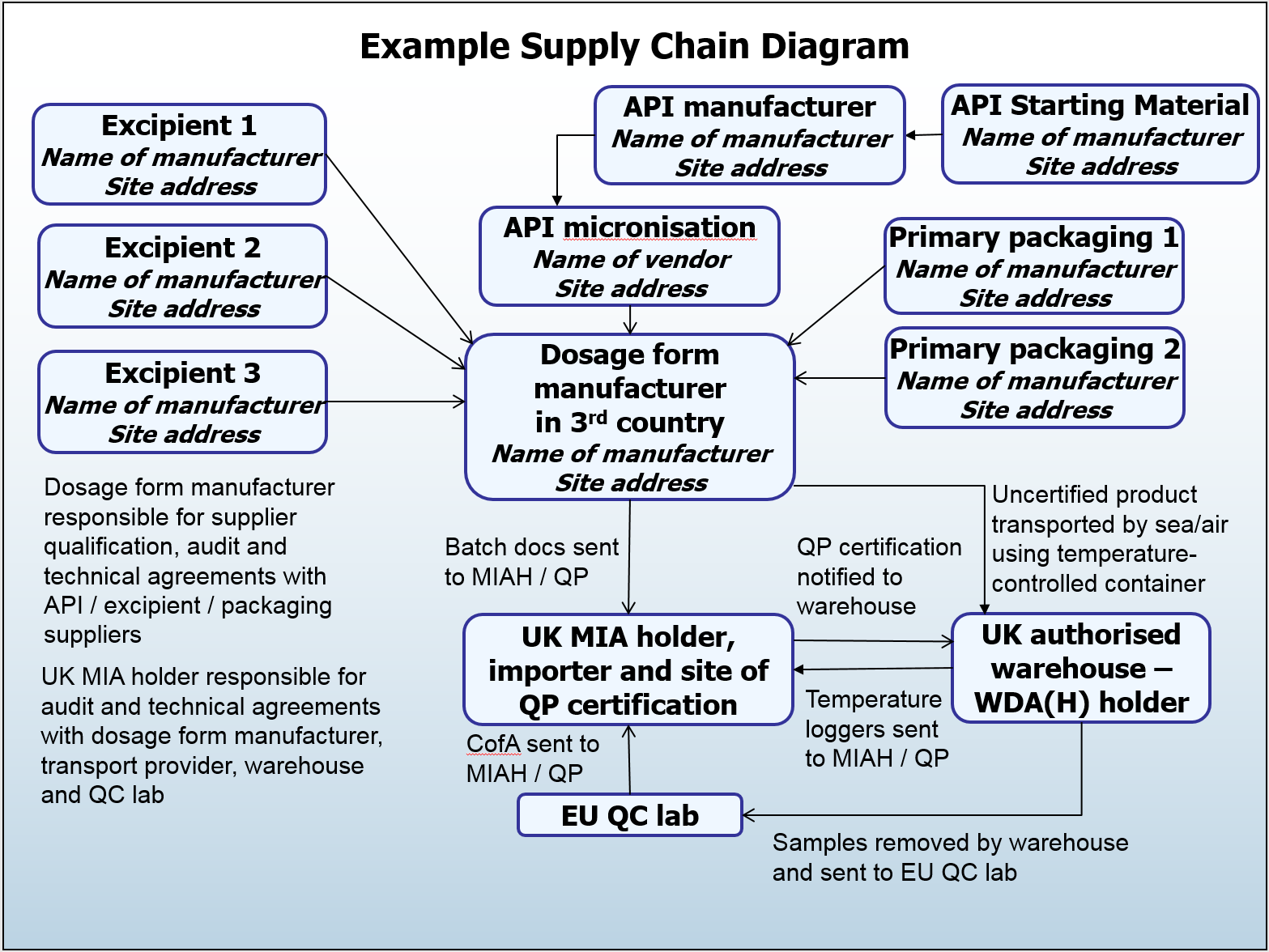
The supply chain can be documented anywhere in the Pharmaceutical Quality System, but it is worthwhile noting that this is a living document which should be maintained to reflect current supply chains. It is preferable for this to be a diagram to aid understanding and avoid any misinterpretation that can be caused by bulky documentation, however this is not mandatory.
Additionally the documentation of the active substance supply chain forms part of the requirements of EU GMP Chapter 1.10 (Product Quality Review), and EU GMP Chapter 5.29 (API supply chain including active substance starting materials).
Part 2 follow-up
In Part 2, my follow-up post will focus on questions relating to the application of EU GMP Annex 16 of QP Certification and batch release of investigational medicinal products.
Don’t miss the next post, sign up to be notified by email when a new post is published on the Inspectorate blog.
Access our guidance on good practice for information on the inspection process and staying compliant.


8 comments
Comment by David Cockburn posted on
I wonder if there is a typo in the example supply chain diagram? Reference is made to MAH but the responsibilities outlines are those of the Manufacturing Authorisation Holder (MIAH).
Comment by Mark Birse posted on
David,
Thank you for your observation
The example we have used in the supply chain diagram for this post is a relatively straightforward one where the MA holder is also the importer and batch cert site (and would therefore also hold an MIA). Likewise, the UK warehouse that is mentioned on the diagram would hold a WDA.
There is a spectrum of business models that we encounter, with multiple permutations of responsibilities and licence holder arrangements, and this is just one example to give an indication of the level of detail expected when documenting supply chains (site addresses, transport arrangements, etc), rather than to focus on the specifics of which licences are held by each party.
I hope this helps.
Mark
Comment by Alain Frix posted on
Any progress on part 2 on QP Certification and batch release of investigational medicinal products?
Comment by Mark Birse posted on
Alain - this is one of the posts we hope to bring you in the coming months
Comment by Jane Self posted on
Hello, is Part 2 likely to be published in the near future please?
Comment by Mark Birse posted on
Jane,
Part 2 was published in August this year and we have now added a link to it in the "You may also be interested in:"box
Comment by Claire Willsher posted on
with the upcoming Brexit in march 2019 will QP batch certification be required if product comes from EU as well as a 3rd country similar to the fact that MA holders must now also reside in the UK?
Comment by Trevor Watson posted on
Claire,
Until the EU-UK negotiations are complete it is not possible to anticipate the future requirements for medicinal products travelling between the UK and EU. The MHRA website will continue to keep stakeholders updated on any developments related to Brexit. The latest post can be found at
https://www.gov.uk/government/news/medicines-and-healthcare-products-regulatory-agency-statement-on-the-outcome-of-the-eu-referendum